Les Neuropathies Optiques Héréditaires (N.O.H.)
Une neuropathie optique est une lésion du ou des nerf(s) optique(s) qui peut aller jusqu’à sa dégénérescence et son atrophie, avec des conséquences graves sur la fonction visuelle. On observe une neuropathie optique dans des maladies aussi diverses que le glaucome ou la sclérose en plaques, mais aussi dans plusieurs maladies génétiques rares.
Parmi les maladies de ce dernier groupe, les neuropathies optiques héréditaires ou N.O.H. sont celles à transmission familiale. Dans les deux principales N.O.H. (L’Atrophie Optique Dominante (maladie de Kjer) et la Neuropathie Optique Héréditaire de Leber), l’atteinte du nerf optique est le seul problème chez la majorité des patients. Il existe cependant des N.O.H. dans lesquelles l’atrophie optique est un élément parmi d’autres dans un tabelau clinique complexe et sévère (syndrome MELAS, syndrome de Wolfram, Ataxie de Friedriech).
OLY se consacre à la lutte contre les Neuropathies Optiques Héréditaires (N.O.H.) et en particulier, mais pas exclusivement, les plus fréquentes :
- L’Atrophie Optique Dominante (ou A.O.D.) ou Maladie de Kjer.
- La Neuropathie Optique Héréditaire de Leber (N.O.H.L)
Ce sont les 2 causes héréditaires les plus fréquentes de cécité, concernant chacune environ 1 individu sur 30 000.
Les N.OH se caractérisent par l’installation sans douleur d’un “brouillard” plus ou moins handicapant au niveau de la vision centrale (qu’on appelle scotome). La vision périphérique peut être plus ou moins épargnée selon les cas.
On peut également être porteur sain, sans jamais développer de symptômes. Les raisons du déclenchement de la maladie sont encore peu connues.
En savoir plus sur le rôle du nerf optique : Oeil pour oeil – C’est pas sorcier – YouTube
Atrophie Optique Dominante (A.O.D.) ou la maladie de Kjer
A retenir
- perte de vision progressive
- se développe dès l’enfance avec des degrés très variables de perte de vision
- transmise par le père ou par la mère
Cette atrophie du nerf optique survient insidieusement et progressivement dans l’enfance. La baisse d’acuité visuelle est très variable, bien que des formes cécitantes soient régulièrement observées.
C’est souvent au cours d’un dépistage à l’école que la maladie est décelée, mais une apparition plus tardive est possible. La déficience visuelle est généralement modérée (l’acuité visuelle varie de 20/80 à 20/120) mais peut varier de légère à sévère.
- La vision des couleurs peut être altérée.
- Le sujet pourra connaître des difficultés avec les couleurs bleu et jaune.
- Des signes extra-oculaires peuvent être présents.
- Dans environ 15 % des cas (forme dite A.O.D. plus), on observe des troubles tels qu’une perte auditive ou d’autres signes neurologiques sévères chez de jeunes adultes, tels qu’une myopathie, des troubles de la coordination, une neuropathie périphérique (engourdissement, fourmillements dans les pieds et/ou les mains) ou une paralysie du mouvement de l’œil.
Diagnostic
Le diagnostic intervient à l’issue de plusieurs examens et recherches :
- un examen ophtalmologique complet
- une recherche d’antécédent familiaux et médicaux
- Des tests génétiques
L’identification prénatale d’une mutation peut être proposée dans les familles lorsque les mutations sont connues, étant entendu que tous les porteurs ne manifesteront pas la maladie.
L’A.O.D. est le plus souvent suspectée chez les enfants présentant une neuropathie optique inexpliquée, surtout si elle est associée à des antécédents familiaux similaires (qui peuvent néanmoins être absents dans 50 % des cas). Le diagnostic, souvent suspecté cliniquement, est confirmé par l’analyse du gène OPA1.
La Maladie de Leber (N.O.H.L.)
A retenir
- prévalence : 1 naissance sur 30 000
- provoque une perte de vision invalidante brutale en quelques semaines ou mois
- chez l’homme : peut se déclencher le plus souvent entre 15 et 30 ans, mais les malades plus jeunes ou plus vieux ne sont pas rares
- chez la femme : peut se déclencher tout au long de la vie
- touche plus souvent les hommes que les femmes (1 femme pour 3 hommes)
- la mutation génétique est transmise par la mère seulement et systématiquement
La maladie a été décrite par Theodor Karl Gustav von Leber (1840-1917) dans une publication de 1871.
NB : Une maladie très similaire à la N.O.H.L. a été décrite ces dernières années qui peut être transmise par les deux parents. Extrêmement rare en Europe occidentale, elle est désignée par LHONAR par l’organisation qui recense les maladies génétiques (OMIM, Online Mendelian Inheritance in Man).
Plus en détail
Histoire naturelle de la maladie
Au début de la maladie (son “déclenchement”), l’acuité visuelle chute brutalement sans douleur ou rougeur. Un premier œil est touché mais le second compense avant d’être atteint lui aussi après quelques semaines. En quelques semaines, la vision est fortement dégradée. La déficience visuelle est sévère et conduit la grande majorité des malades sous le seuil légal de cécité (acuité visuelle inférieure à 1/20). C’est la phase aiguë de la maladie.
Après la phase aiguë commence la phase chronique de la maladie, qui va durer toute la vie. En général, la vision reste stable, même s’il arrive qu’elle s’améliore un peu après quelques mois. Dans de rares cas, et pour des raisons inconnues, la vision s’améliore significativement 18 à 24 mois après l’installation de la maladie, sans pour autant lever la déficience visuelle. Dans la grande majorité des cas, les malades restent sous le seuil légal de cécité.
Il persiste une tache aveugle, plus ou moins étendue, au centre du champ visuel.
La personne touchée utilisera un regard excentré pour percevoir. La sensibilité aux contrastes est altérée. Les objets sont mieux perçus lorsqu’ils sont présentés à contraste maximal, c’est à dire noirs sur fond blanc. La perception des couleurs est altérée ou perdue (daltonisme ou dyschromatopsie).
Diagnostic
Le diagnostic de la neuropathie optique héréditaire de Leber combine différents éléments cliniques, génétiques et d’imagerie, à savoir :
- un examen ophtalmologique complet : image du fond d’œil, évaluation du champ visuel, tomographie par cohérence optique (O.C.T)
- une recherche d’antécédent familiaux et médicaux
- l’exclusion de causes infectieuses, compressives (tumeur) ou dues à des carences ou intoxications
- la recherche de mutations génétiques
Seul le test génétique permet de poser le diagnostic avec certitude.
Les Protocoles Nationaux de Diagnostic et de Soins sont disponibles ici pour les professionnels de la santé
Causes
La N.O.H.L est due à des mutations génétiques qui affectent l’ADN mitochondrial maternel : ce sont des mutations primaires : elles sont requises, mais pas suffisantes pour déclencher la maladie.
Selon les études, la N.O.H.L. se déclenche chez 20-50 % des hommes et 5-30 % des femmes porteurs d’une mutation primaire. Les porteurs asymptomatiques (ou porteurs sains) des mutations sont donc nombreux : ce sont les membres non malades de la lignée maternelle d’une famille contenant au moins un malade.
Le tabagisme et l’alcoolisation aigüe massive sont les facteurs de risques majeurs et démontrés du déclenchement de la N.O.H.L..
Certaines substances ou situations pourraient accélérer ou aggraver les symptômes : les toxines, les polluants environnementaux, le stress ou encore les situations de carences.
Mécanisme de la maladie
Les mutations de la N.O.H.L. affectent des gènes importants pour le fonctionnement de la mitochondrie, ce qui provoque la diminution de la production d’énergie, une augmentation du « stress oxydatif » (l’excès de certaines molécules oxygénées qui abîment la cellule) qui peut provoquer la mort de la cellule. Or, de toutes les cellules du corps, les neurones ganglionnaires de la rétine, aussi appelés cellules ganglionnaires de la rétine (CGR), sont parmi les plus demandeuses d’énergie et les plus fragiles.
La rétine tapisse l’arrière de l’œil et contient une multitude de cellules connectées, distribuées en couches superposées et qui convertissent la lumière en signaux biologiques puis électrophysiologiques dans le cerveau (Figure 1).
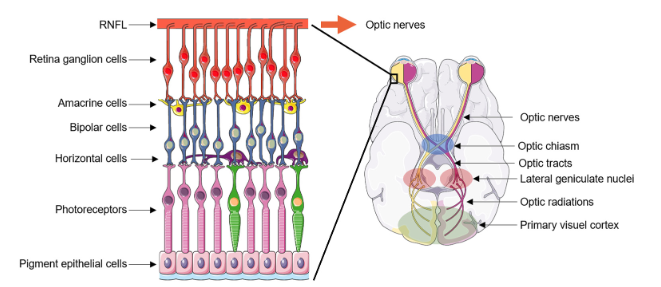
Les CGRs tapissent la surface de la rétine, à partir de laquelle ils projettent des extensions de leur corps cellulaire appelés axones. Leurs axones forment une couche qui recouvre la rétine, appelée la couche de fibres neurales de la rétine (RNFL). C’est elle dont on mesure l’épaisseur lors de l’examen OCT. Les axones se regroupent ensuite pour former les nerfs optiques. Les nerfs optiques des deux yeux se rejoignent au niveau du chiasma optique, où leurs fibres se croisent pour former les voies optiques, qui poursuivent la transmission de l’information visuelle vers les régions du cerveau impliquées dans la vision.
A cause de l’organisation anatomique de la rétine, les CGRs sont des neurones très dépendants de la ressource en énergie et très fragiles : leurs axones doivent être courbés et ils ne sont pas protégés comme les autres neurones du corps par la gaine dite « de myéline » sur toute leur longueur. Les CGRs sont donc particulièrement impactés par la dysfonction mitochondriale lors du déclenchement de la maladie. C’est leur mort qui provoque l’atrophie optique et la déficience visuelle. Dans la N.O.H.L., la dégénérescence touche préférentiellement les petites fibres qui assurent la vision centrale.
Comme le montre la tomographie par cohérence optique (OCT), chez les patients atteints de N.O.H.L., la RNFL gonfle au cours des premiers mois avant de s’atrophier et enfin de se stabiliser dans la phase chronique. L’examen histopathologique de sections prélevées sur des patients décédés a révélé une perte importante de CGRs et un amincissement sévère de la RNFL et, au niveau du nerf optique, une perte massive de fibres, une réduction de la section transversale du nerf et des vestiges de neurodégénérescence.
Recommandations pour les patients et porteurs sains N.O.H.L.
L’environnement joue un rôle clé dans le déclenchement et l’expression de la neuropathie optique héréditaire de Leber (N.O.H.L.). Sur la base de la bibliographie et de notre revue publiée dans Biomedecines avec le Toulouse Neuroimaging Center, voici des recommandations pour les patients et porteurs sains de mutations N.O.H.L. :
Adopter une alimentation équilibrée : privilégier un régime riche en fruits, légumes, fibres, poissons, huiles végétales et pauvre en sucres raffinés, comme par exemple le régime méditerranéen. Le régime cétogène n’est pas encore assez connu pour être suivi sans supervision médicale. Privilégiez les produits « bio ».
Éviter les facteurs de risque : ne jamais fumer (ni tabac ni cannabis, même sous la forme de CBD ; le vapotage est une bonne solution de sevrage), éviter de consommer de l’alcool en excès, ne pas s’exposer à des solvants ou des composés organiques volatils (le poppers en est un) ou des fumées quelle que soit la nature du feu.
Pratiquer une activité physique régulière : l’exercice physique, notamment l’endurance, favorise la santé mitochondriale et réduit le stress oxydatif.
Surveiller les carences nutritionnelles : faire vérifier vos taux de vitamine B12 et vitamine B3 car des déficits ont été observés chez certains patients.
Traitements, médicaments et compléments :
- Toujours signaler aux médecins si vous êtes patient ou porteur sain ou s’il existe des antécédents dans votre famille
- Informer tout particulièrement le praticien avant une anesthésie ou pour traiter l’épilepsie ou l’arythmie cardiaque
- Être vacciné contre la tuberculose et rester à jour (le traitement anti-tuberculose peut déclencher la N.O.H.L.)
- Eviter les conduites à risques et se protéger du VIH (le traitement anti-rétroviral peut déclencher la N.O.H.L.)• Respecter les doses indiquées pour les compléments alimentaires ; même si un grand nombre sont potentiellement bénéfiques (Coenzyme Q10, anti oxydants, taurine), leur efficacité n’est pas démontrée.
En résumé, la prévention repose principalement sur l’hygiène de vie, l’évitement des toxiques et la correction des carences, en concertation avec une équipe médicale spécialisée.Source et pour en savoir plus : un article (en anglais, accès libre) sur les facteurs environnementaux et la N.O.H.L. publié par OLY et le groupe de Stéphane Chavanas, conseiller scientifique d’Ouvrir les yeux, que vous trouverez ici.
Comment se transmettent les N.O.H.
Gènes et mitochondries
Les gènes sont les unités fondamentales de l’hérédité. Ils sont portés par l’ADN, une longue molécule présente dans le noyau de chaque cellule. C’est un code de trois milliards de signes qu’on appelle le génome, propre à chaque individu. Le génome contient vingt mille gènes, qui donnent aux cellules du corps les instructions pour fabriquer toutes les protéines du corps. Le génome est réparti en 23 chromosomes, chacun présent en deux exemplaires. Ces chromosomes, et donc les gènes qu’ils portent, sont transmis à la descendance par le père et par la mère : un exemplaire du père, un exemplaire de la mère. Deux chromosomes parmi les 23 font exception : ce sont les chromosomes sexuels. La femme a deux chromosomes X et transmettra l’un ou l’autre, l’homme a un chromosome X et un chromosome Y et transmettra l’un ou l’autre.
Chaque cellule contient aussi les mitochondries, en dehors du noyau. Les mitochondries sont des petites entités en forme de gnocchis d’un millionième de mètre, très dynamiques, qui passent leur temps à fusionner ou à se séparer les unes et les autres. Chaque neurone en contient plusieurs centaines, jusqu’à 2000. Les mitochondries produisent notre énergie. Elles jouent également un rôle essentiel dans le métabolisme, la détoxification, la survie de la cellule face à une agression, et dans l’immunité. Leur fonction est vitale. Les mitochondries contiennent une petite molécule d’ADN présente en une dizaine d’exemplaires : c’est l’ADN mitochondrial. Cet ADN ne contient que 16000 signes mais porte des gènes indispensables à la vie : c’est le génome mitochondrial. Ce génome mitochondrial est transmis à la descendance par la mère seulement.
La fécondation
Lors de la fécondation, un spermatozoïde du père pénètre dans l’ovocyte de la mère. La moitié de l’ADN nucléaire du père et la moitié de l’ADN nucléaire de la mère se mélangent alors pour former le génome nucléaire de l’enfant à naître. L’enfant hérite donc d’une version maternelle et d’une version paternelle de chaque gène du génome nucléaire.
Les mitochondries du spermatozoïde ne sont pas transmises à l’œuf. Seules les mitochondries de l’ovocyte sont conservées dans l’œuf. Par conséquent, seul le génome mitochondrial de la mère est transmis à l’enfant à naître. L’enfant hérite seulement de la version maternelle des gènes mitochondriaux.
Il arrive qu’un gène subisse une mutation qui perturbe son fonctionnement au point de provoquer une maladie. Cette mutation génétique risque alors d’être transmise aux enfants.
Qu’en est-il dans le contexte des N.O.H. ?
Atrophies optiques dominantes (A.O.D. ou maladie de Kjer)
La maladie de Kjer est causée par une mutation génétique dans l’ADN nucléaire. Elle peut être transmise par le père ou par la mère.
Cette maladie est dite « dominante », ce qui signifie que si l’un ou l’autre des parents est porteur de la mutation, l’enfant a 50% de risques d’en hériter, quel que soit son sexe.
Cette forme n’est pas liée au sexe : filles et garçons sont également concernés.
Neuropathie optique héréditaire de Leber (N.O.H.L.)
La neuropathie optique héréditaire de Leber est causée par une mutation génétique dans l’ADN mitochondrial.
Elle est donc transmise uniquement par la mère. Si une mère est porteuse de la mutation, elle a 100% de risques de la transmettre à ses enfants.
Une femme porteuse de la mutation, qu’elle soit malade ou pas, transmet la mutation à tous ses enfants, garçons comme filles.
Un homme porteur de la mutation, qu’il soit malade ou pas, ne transmet pas la maladie à ses enfants.
Cependant, l’enfant ne sera pas forcément malade : il peut être porteur sain toute sa vie.
Atrophies optiques récessives
Ici, la maladie apparaît seulement si l’enfant reçoit deux copies mutées du gène : un exemplaire muté de sa mère, un exemplaire muté de son père.
Les parents peuvent eux-mêmes être porteurs sains.
Les parents, non malades, sont tous deux porteurs d’un exemplaire du gène muté.
L’enfant doit hériter de la mutation des deux côtés pour être malade.
Si aucun des parents est malade, la grossesse comporte un risque de 1 sur 4 (25 %) d’avoir un enfant malade, quel que soit le sexe.
Si l’un des parents est malade, la grossesse comporte un risque de 3 sur 4 (75 %) d’avoir un enfant malade, quel que soit le sexe.
Atrophies optiques liées à l’X (récessif lié à l’X)
Dans ce cas, la mutation se trouve sur le chromosome X qui est un chromosome sexuel : les femmes en ont deux copies, alors que les hommes n’en ont qu’une.
Les femmes peuvent être « conductrices » (porteuses sans être malades), car elles ont deux chromosomes X : le chromosome sain compense l’autre muté chez elles.
Elles ont une chance sur 2 de transmettre le X muté à leur descendance, que ce soit une fille ou un garçon. Dans ce cas, une fille sera à son tour conductrice et un garçon sera atteint.
Un homme atteint ne transmet pas la maladie à ses fils, mais il transmet son unique chromosome X, qui est muté, à toutes ses filles qui de facto deviennent toutes conductrices.
Grossesse : Peut-on savoir si un enfant à naître est porteur d’une mutation
Le diagnostic génétique anténatal permet aujourd’hui d’identifier précocement chez l’embryon ou le fœtus la présence de mutations responsables de maladies génétiques héréditaires. En cas de grossesse, ce diagnostic donne aux familles concernées la capacité de choisir une prise en charge adaptée et informée. Cela permet de :
- réduire l’incertitude pour les parents ayant un antécédent familial de neuropathie optique.
- • préparer une prise en charge médicale et psychosociale de l’enfant à naître, si la mutation est détectée chez lui
- • offrir, dans certains cas spécifiques, la possibilité d’envisager une interruption médicale de grossesse.
Il y a une condition préalable : la mutation dans la famille doit être connue.
Quand il est possible, le diagnostic anténatal est obtenu grâce à l’analyse génétique d’un prélèvement de villosités choriales. Le prélèvement des villosités choriales consiste à prélever un échantillon minuscule du placenta à l’aide d’une aiguille ou d’un tube fin, en passant par le col de l’utérus (ou par le ventre).
Le diagnostic anténatal est envisageable pour les atrophies optiques dominantes ou récessives si la mutation familiale (par exemple sur le gène OPA1) a été identifiée.
Pour la N.O.H.L., à cause de la transmission mitochondriale de 100% de l’ADN muté à l’enfant dans la grande majorité des cas, le diagnostic anténatal n’a d’intérêt que dans des cas très rares. De surcroît, il pose de grandes difficultés d’interprétation et d’éthique, car tous les enfants porteurs ne développent pas la maladie.
Dans tous les cas, cette démarche nécessite une orientation vers une consultation de génétique et une indication médicale rigoureuse.